Safety in Dravet syndrome
FINTEPLA has an established safety profile1
- Despite initial weight loss, most patients resumed the expected measured increases in weight during the OLE study
- Growth and weight of pediatric patients should be carefully monitored during treatment with FINTEPLA, and dose modifications should be considered if a decrease in weight is observed
- Somnolence, sedation, and lethargy may diminish with continued treatment
- Other central nervous system depressants, including alcohol, could potentiate these effects of FINTEPLA
- Monitor patients for somnolence and sedation, and advise them not to drive or operate machinery
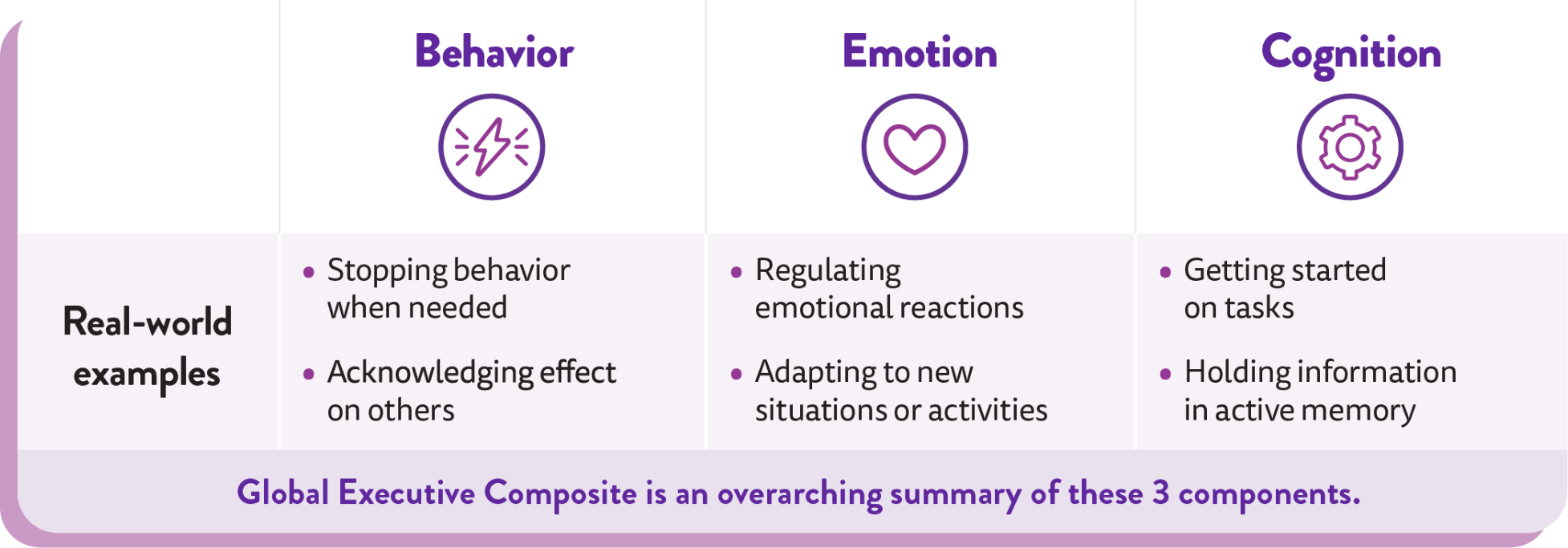
Evaluation of behavior, emotion, and cognition, as measured by the BRIEF®
The Behavior Rating Inventory of Executive Function (BRIEF) is a validated tool in neurology and is the most widely used rating scale for assessing everyday behavior, emotion, and cognition (executive function) in clinical trials. BRIEF was used as a safety endpoint in FINTEPLA studies.2,3
BRIEF evaluation at a glance2
Aggression has been identified during postapproval use of FINTEPLA. These voluntary reports from a population of uncertain size prevent reliable estimates of frequency or causality.
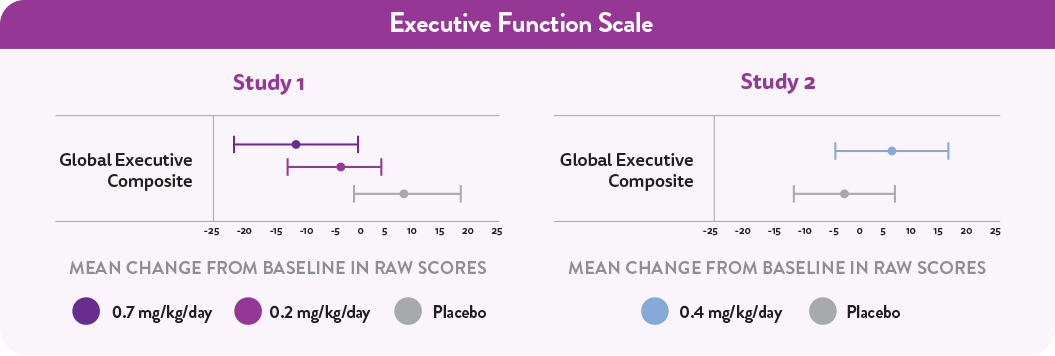
Executive Function scale (includes behavior, emotion, and cognition)3,4
References
1. FINTEPLA (fenfluramine) oral solution: U.S. prescribing information. Smyrna, GA: UCB, Inc.; 2. Bishop KI, Isquith PK, Gioia GA, et al. Improved everyday executive functioning following profound reduction in seizure frequency with fenfluramine: analysis from a phase 3 long-term extension study in children/young adults with Dravet syndrome. Epilepsy Behav. 2021;121(pt A):109024. doi:10.1016/j.yebeh.2021.108024; 3. Data on file. UCB, Inc.; 4. Lagae L, Sullivan J, Knupp K, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. 2019;394(10216):2243-2254. doi:10.1016/S0140-6736(19)32500-0
Important Safety Information
See moreBOXED WARNING: VALVULAR HEART DISEASE and PULMONARY ARTERIAL HYPERTENSION
- FINTEPLA can cause valvular heart disease and pulmonary arterial hypertension.
- Echocardiogram assessments are required before, during, and after treatment with FINTEPLA.
- FINTEPLA is available only through a restricted program called the FINTEPLA REMS.
CONTRAINDICATIONS
FINTEPLA is contraindicated in patients with hypersensitivity to fenfluramine or any of the excipients in FINTEPLA and with concomitant use, or within 14 days of the administration, of monoamine oxidase inhibitors because of an increased risk of serotonin syndrome.
WARNINGS AND PRECAUTIONS
Valvular Heart Disease and Pulmonary Arterial Hypertension (see Boxed Warning): FINTEPLA can cause valvular heart disease (VHD) and pulmonary arterial hypertension (PAH). Although no patients receiving FINTEPLA developed VHD or PAH in clinical trials for DS and LGS of up to 3 years in duration, cases of VHD and PAH have been reported during use of FINTEPLA in the postmarketing setting. Because of this risk, cardiac monitoring is required prior to starting treatment, during treatment, and after treatment with FINTEPLA concludes. Cardiac monitoring via echocardiogram can identify evidence of VHD and PAH prior to a patient becoming symptomatic, aiding in early detection of these conditions.
Monitoring: Prior to starting treatment, patients must undergo an echocardiogram to evaluate for VHD and PAH. Echocardiograms should be repeated every 6 months, and once 3-6 months post treatment with FINTEPLA.
The prescriber must consider the benefits versus the risks of initiating or continuing treatment with FINTEPLA if any of the following signs are observed via echocardiogram: valvular abnormality or new abnormality; VHD indicated by mild or greater aortic regurgitation or moderate or greater mitral regurgitation, with additional characteristics of VHD (e.g., valve thickening or restrictive valve motion); PAH indicated by elevated right heart/pulmonary artery pressure (PASP >35 mm Hg).
FINTEPLA REMS (see Boxed Warning): FINTEPLA is available only through a restricted distribution program called the FINTEPLA Risk Evaluation and Mitigation Strategy (REMS). Prescribers must be certified by enrolling in the FINTEPLA REMS. Prescribers must counsel patients receiving FINTEPLA about the risk of VHD and PAH, how to recognize signs and symptoms of VHD and PAH, the need for baseline (pretreatment) and periodic cardiac monitoring via echocardiogram during FINTEPLA treatment, and cardiac monitoring after FINTEPLA treatment.
Decreased Appetite and Decreased Weight: FINTEPLA can cause decreases in appetite and weight. Decreases in weight appear to be dose related. Approximately half of the patients with LGS and most patients with DS resumed the expected measured increases in weight during the open-label extension studies. Weight should be monitored regularly during treatment with FINTEPLA, and dose modifications should be considered if a decrease in weight is observed.
Somnolence, Sedation, and Lethargy: FINTEPLA can cause somnolence, sedation, and lethargy. Other central nervous system depressants, including alcohol, could potentiate these effects of FINTEPLA. Prescribers should monitor patients for somnolence and sedation and should advise patients not to drive or operate machinery until they have gained sufficient experience on FINTEPLA to gauge whether it adversely affects their ability to drive or operate machinery.
Suicidal Behavior and Ideation: Antiepileptic drugs (AEDs), including FINTEPLA, increase the risk of suicidal thoughts or behaviors in patients taking these drugs for any indication. Patients treated with an AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behaviors, or any unusual changes in mood or behavior.
Withdrawal of Antiepileptic Drugs: As with most AEDs, FINTEPLA should generally be withdrawn gradually because of the risk of increased seizure frequency and status epilepticus. If withdrawal is needed because of a serious adverse reaction, rapid discontinuation can be considered.
Serotonin Syndrome: Serotonin syndrome, a potentially life-threatening condition, may occur with FINTEPLA, particularly during concomitant administration of FINTEPLA with other serotonergic drugs, including, but not limited to, selective serotonin-norepinephrine reuptake inhibitors (SNRIs), selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants (TCAs), bupropion, triptans, dietary supplements (e.g., St. John’s Wort, tryptophan), drugs that impair metabolism of serotonin (including monoamine oxidase inhibitors [MAOIs], which are contraindicated with FINTEPLA), dextromethorphan, lithium, tramadol, and antipsychotics with serotonergic agonist activity. Patients should be monitored for the emergence of signs and symptoms of serotonin syndrome, which include mental status changes, autonomic instability, neuromuscular signs, and/or gastrointestinal symptoms. If serotonin syndrome is suspected, treatment with FINTEPLA should be stopped immediately and symptomatic treatment should be started.
Increase in Blood Pressure: FINTEPLA can cause an increase in blood pressure. Rare cases of significant elevation in blood pressure, including hypertensive crisis, have been reported in adult patients treated with fenfluramine, including patients without a history of hypertension. In clinical trials for DS and LGS of up to 3 years in duration, no pediatric or adult patient receiving FINTEPLA developed hypertensive crisis. Monitor blood pressure in patients treated with FINTEPLA.
Glaucoma: Fenfluramine can cause mydriasis and can precipitate angle closure glaucoma. Consider discontinuing treatment with FINTEPLA in patients with acute decreases in visual acuity or ocular pain.
ADVERSE REACTIONS
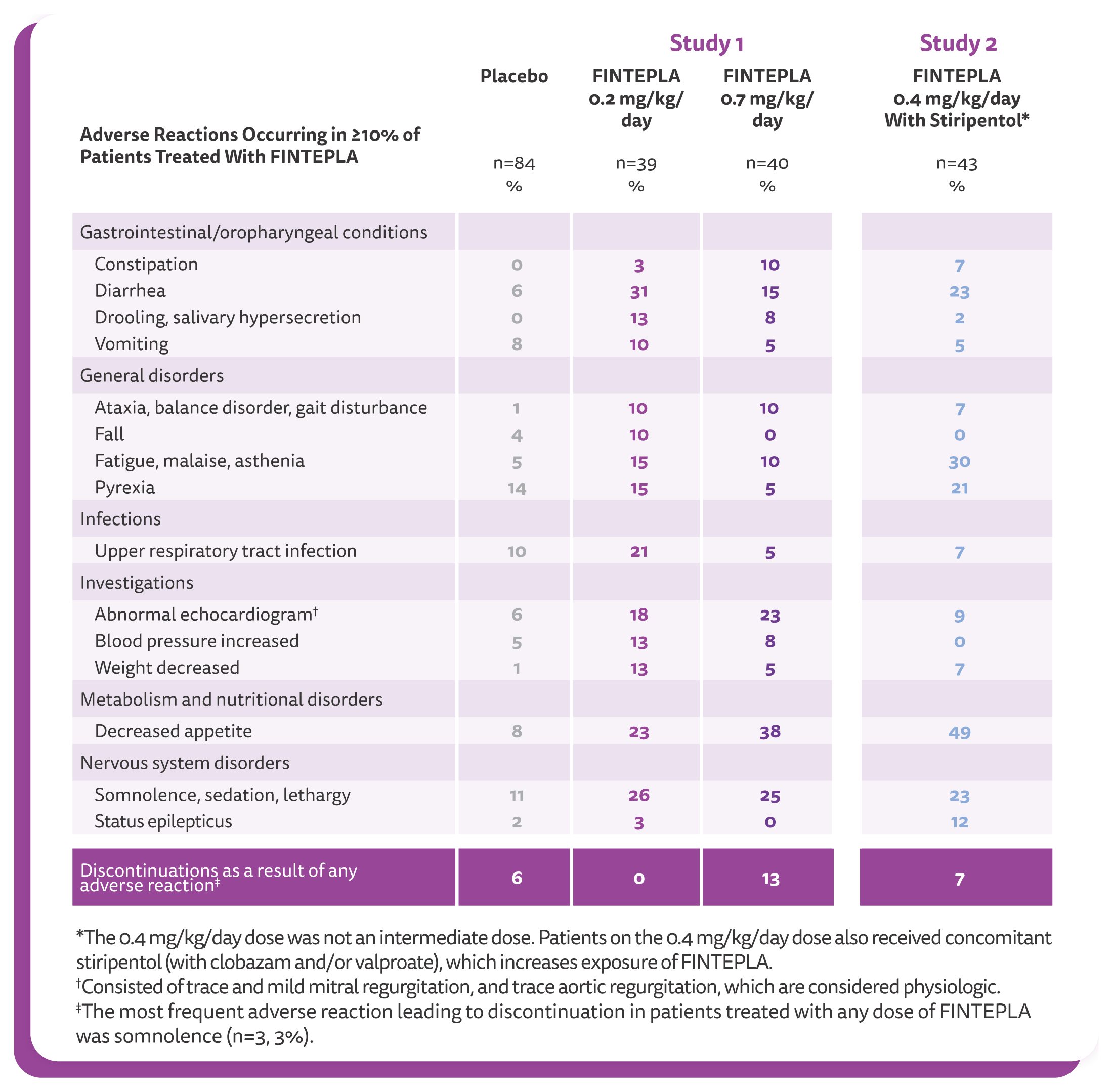
The most common adverse reactions observed in DS studies (incidence at least 10% and greater than placebo) were decreased appetite; somnolence, sedation, lethargy; diarrhea; constipation; abnormal echocardiogram; fatigue, malaise, asthenia; ataxia, balance disorder, gait disturbance; blood pressure increased; drooling, salivary hypersecretion; pyrexia; upper respiratory tract infection; vomiting; decreased weight; fall; status epilepticus.
The most common adverse reactions observed in the LGS study (incidence at least 10% and greater than placebo) were diarrhea; decreased appetite; fatigue; somnolence; vomiting.
To report SUSPECTED ADVERSE REACTIONS, contact UCB, Inc. at 1‑844-599-2273 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Please see full Prescribing Information, including Boxed Warning, for additional Important Safety Information.